

簡介

衛生福利部 醫療器材品質管理系統準則QMS
醫療器材品質管理法已於110年5月1日正式施行,展現了政府對於醫療器材品質管理的重視,隨著醫療器材管理法的施行,相關的施行細則、醫療器材品質系統要求與檢驗、醫療器材製造許可制度等,均有大幅度的變化。
對於國產醫療器材業者在醫療器材管理法第22條中規定:醫療器材製造業者應建立醫療器材品質管理系統,就場所設施、設備、組織與人事、生產、品質管制、儲存、運銷、客戶申訴及其他事項予以規範,並應符合品質管理系統準則。
醫療器材製造業者依前項準則規定建立醫療器材品質管理系統,並報中央主管機關檢查合格取得製造許可後,始得製造。但經中央主管機關公告之品項,免取得製造許可。
醫療器材品質管理系統準則,主要是參照ISO 13485:2016之內容訂定。因此科建顧問於輔導醫療器材製造廠時,均會為客戶訂做符合國際標準與國內法規的品質管理制度,協助客戶開拓內、外銷通路時,滿足市場對於醫療器材品質管理的要求,拓展商機。
適用對象
國內醫療器材製造業
預期效益
符合國內醫療器材法規之要求,成為合格醫療器材製造廠商。
對醫療器材做適當的風險管理,以確保醫療器材產品安全性。
跨越醫療器材之入門障礙,作為申請查驗登記與銷售的準備。
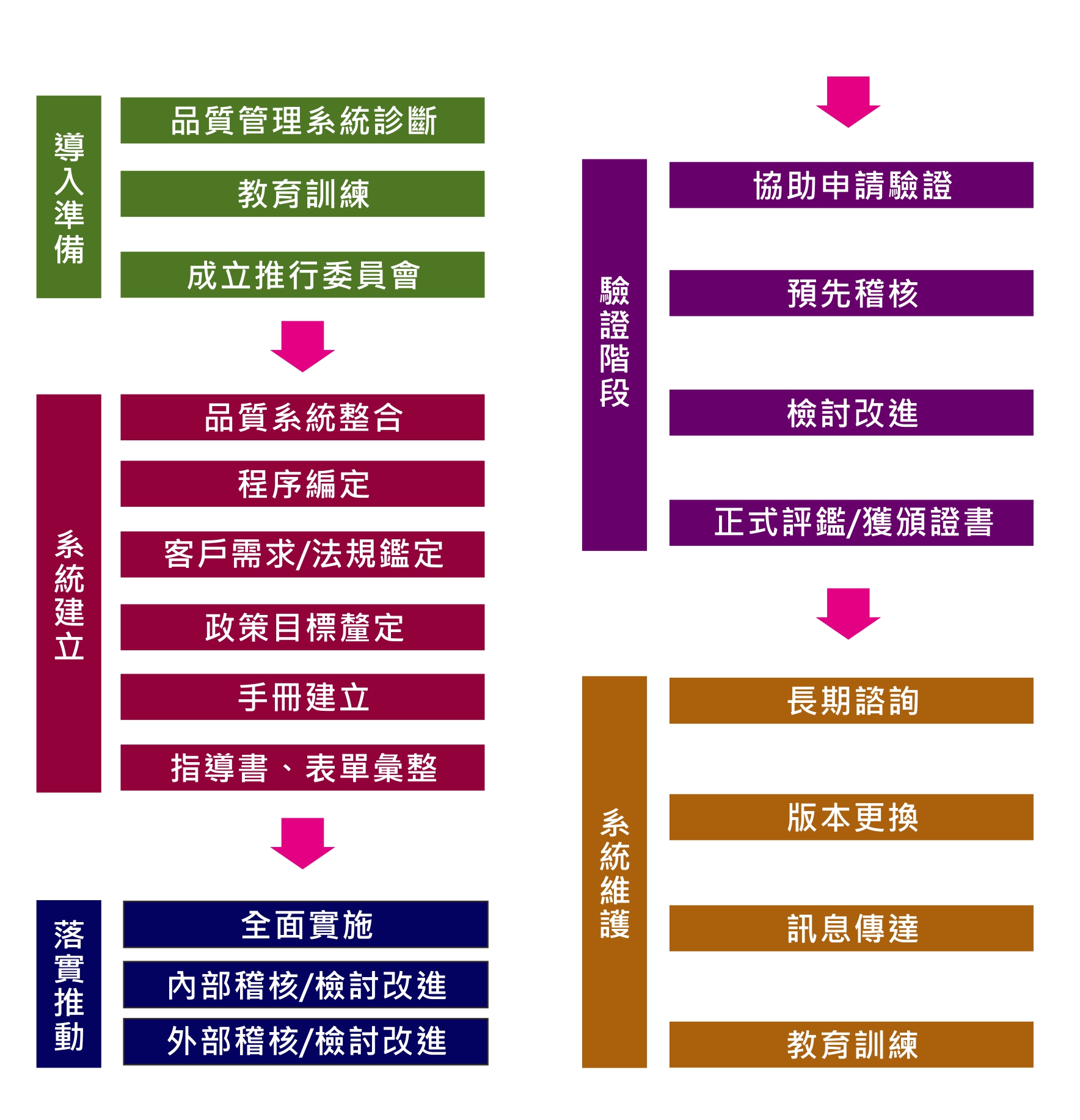
輔導流程
REPORT / 新聞稿
ARTICLES / 文章分享
-
2010.12.16
淺論醫療器材優良製造規範(GMP)之設計開發
隨著時代進步,醫療器材產品的技術日新月異,不但種類愈來愈多樣,功能也愈趨進步,這些都是醫療器材不斷研發創新的結果,我們國內的醫療器材優良製造規範對於導入之醫療器材業者原則上都要求要做設計開發,以下淺論醫療器材在設計開發階段之相關要求。
一、設計開發規劃階段
在此條款中業者應將設計活動區分為數個階段,各階段可由業者自行定義,且毋須依條文順序作區分,各階段內應執行哪些設計活動?各活動歸屬何種執行單位?亦需予以界定。在規劃設計流程時,應注意實際執行面之順序以及各單位間的分工,例如:某電子產品設計專案應指派專案負責人(project leader),專案成立後又分為軟體、硬體、機構三個開發專案,各專案由何處分道揚鑣、何處匯合河流、各由何單位負責,都須加以規定,設計活動規劃妥當後始可進行。各項設計活動關係請參見附圖:設計開發關聯圖。
二、設計開發輸入
此階段最主要的工作是定義醫療器材的『功能』與『預期用途』。研發人員應針對醫療器材定義其預期醫療效能與使用方式,並將安規之要求輸入產品內。設計輸入之要求應執行設計審查,以確認其適當、明確且不相衝突,另外,此時應執行醫療器材風險管理活動,若經評估為不可接受之風險則須納入設計輸入階段。
三、設計開發輸出
醫療器材產品設計各階段均有其設計輸出,每個階段的輸出多為表單記錄。由於設計活動各階段多半有設計審查,以作為是否進入下個階段的參考依據,故各階段的設計審查作業記錄即為該階段之設計輸出。
四、設計開發審查
設計活動各階段的結果均須召集相關單位代表實施評估,以決定設計結果是否符合該階段之要求,若結果符合則進入下個設計階段;若不符合則應提出檢討以確認產品的潛在問題,該問題若可以解決則應提出設計變更。
五、設計開發驗證
設計驗證作業主要目的在於確認設計輸出的規格,經實際檢測後結果是否符合,此項作業應注意所有開立之規格均須加以查證,且醫療器材安規的檢驗亦應納入設計驗證活動之一環。
六、設計開發確認
設計確認作業的主要目的在確認設計的醫療器材產品是否符合所定義之預期用途,故設計確認作業通常於客戶端進行。此項目在醫療器材優良製造規範上應注意下列要求:
1.若醫療器材必須於客戶端執行組裝,在未完成組裝前,不能視為已交貨。
2.若醫療器材必須作臨床評估,則臨床評估應視為設計確認的一部分。廠商若於國內實施臨床評估,則應先 提出臨床評估計劃經主
管機關審核後始得執行。3.廠商必須於設計驗證完成且通過(PASS)後始得實施臨床評估,若設計驗證與安規檢測未通過即實施臨床評估,一般代施查核機構
較不接受(會開立主要缺失)。七、設計變更
醫療器材之設計變更與一般產品一樣,須經核准後始得起動,以昭謹慎。執行變更時應明列變更前後之差異、變更原因、變更驗證等,以確保變更的正確性,減少變更次數。
綜合上述各項條文說明可得知,醫療器材優良製造規範對於設計開發活動之要求較ISO 9001更為嚴謹,其宗旨在於藉由實施設計活動使醫療器材的功能與效益提至最高,產品風險降至最低,以俾物盡其用,造福人群。
Q & A / 問答集
-
請問醫療器材優良製造規範於驗證申請上有何要求?
申請醫療器材優良製造規範的工廠,必須符合國內醫療器材工廠設廠標準,並取得工廠登記證以及藥商製造許可執照,為符合上述要求,廠商的工廠必須位於工業用地,另外由於本規範為優良作業規範,故申請廠商必須要有醫療器材生產、製造、組裝或加工等作業,不能僅為銷售通路業者。上述登記證應向地方縣市政府衛生局提出申請。
-
請問醫療器材優良製造規範於產品方面有何要求?
醫療器材優良製造規範所驗證的產品必須為已經設計開發完成,並得以量產上市之醫療器材。在設計過程中產品之安規測試必須測試通過,以確認其安全性,若尚未設計完成的產品,無法申請醫療器材優良製造規範之驗證。
-
請問醫療器材優良製造規範與ISO 13485要求有何差別?
基本上醫療器材優良製造規範是給國內醫療器材廠商應用的,ISO 13485則是各國醫療器材業者皆可導入,且醫療器材優良製造規範要求必須要有設計開發階段,ISO 13485則允許排除設計開發。另外,醫療器材優良製造規範之導入廠商必須完成產品之製造、組裝作業,ISO 13485則不限成品或半成品,且不限製造與否,若僅做設計或銷售通路者亦可導入ISO 13485。
-
請問醫療器材優良製造規範驗證缺失如何分類?
國內醫療器材優良製造規範驗證之缺失分為主要缺點與次要缺點,正式驗證時若有主要缺點一項,或次要缺點超過十項,皆會造成驗證失效。
-
請問醫療器材優良製造規範驗證時產品如何分級分類?
國內醫療器材分類與分級是由衛生署執行分級分類,並公告於網站上,若廠商製造之醫療器材過於新穎,衛生署尚未分類分級者,可填寫醫療器材列管查核申請表向衛生署提出分類申請。